
GROMACS is a versatile package to perform molecular dynamics, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles. Further details are described at www.gromacs.org/About_Gromacs.
GROMACS is a popular HPC application and hence one that is interesting to characterize for performance as well as to analyze how it uses my reference architectures. Following are some more specific postings I made when I was setting up the gromacs installation.
After I had the executables built, the next step was to find example workloads to run and measure. These have additional detail on pages below.
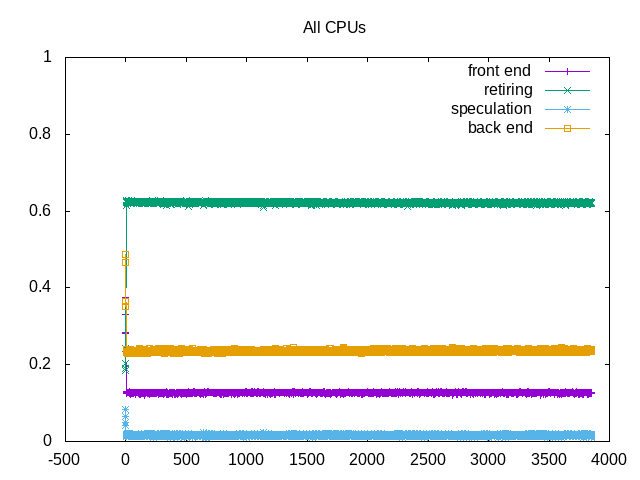 About this graph
About this graph
This example shows topdown metrics for lysozyme md stage.
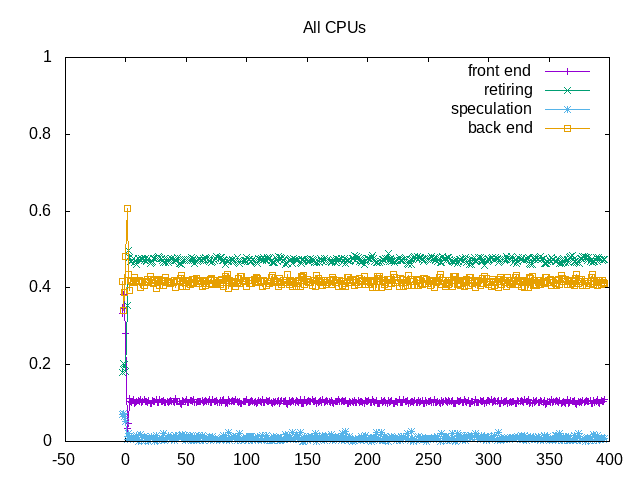 About this graph
About this graph
This example shows topdown metrics for ion channel workload (more backend memory stalls and lower IPC)
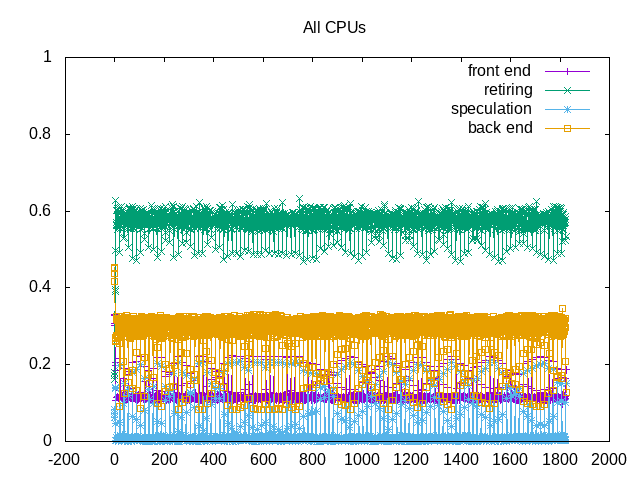 About this graph
About this graph
This example shows topdown metrics for the lignocellulose workload (intermediate level of backend memory stalls and slightly higher IPC; also some more noise in the measurement).
In addition to this characterization, it is also useful to note that gromacs documentation includes a section on performance. Of particular note is that gromacs uses both mpi and openmp to partition the work both between nodes and also within multiple cores of a node. Potentially useful to use these configurations to more accurately map gromacs computations in a multi-node environment.